医械资讯周刊 | 2024年8月第一期
 90
901、国家药监局关于印发医疗器械经营质量管理规范现场检查指导原则的通知
新修订的《医疗器械经营质量管理规范》(以下简称《规范》)自2024年7月1日起施行。为规范和指导医疗器械经营质量管理规范现场检查工作,国家药监局组织制定了《医疗器械经营质量管理规范现场检查指导原则》(以下简称《指导原则》),现予印发。
本《指导原则》适用于药品监督管理部门依据《规范》,对医疗器械经营企业经营许可(含变更和延续)现场核查,或者经营备案后的现场检查,以及其他各类监督检查。检查过程中,医疗器械经营企业可以根据其经营方式、经营范围、经营品种等特点,确定合理缺项项目,并书面说明理由,由药品监督管理部门的检查组予以确认。
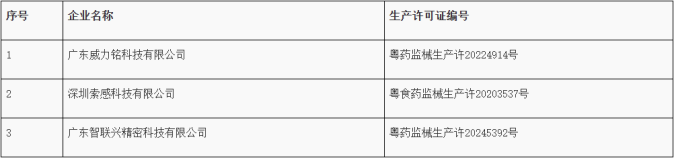
2、广东省药品监督管理局关于3家医疗器械生产企业恢复生产的通告
经广东省药品监督管理局组织复查,以下3家医疗器械生产企业已完成缺陷项目整改,符合《医疗器械生产质量管理规范》相关规定恢复生产。
3、关于公开征求对免于临床评价医疗器械目录意见的通知
为贯彻落实中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),进一步规范医疗器械临床评价工作,扩大免于临床评价医疗器械目录(以下简称器械豁免目录)范围,器审中心启动了2024年度器械豁免目录的制修订工作。目前器审中心形成了《建议新增和修订的免于临床评价医疗器械目录(2024年征求意见稿)》(见附件1),即日起在中心网站公开征求意见,各有关单位对2023年发布的器械豁免目录提出建议新增和修订的意见。相关意见或建议,请填写意见反馈表,以“2024年器械豁免目录意见反馈”为邮件主题至邮箱:huangyl@cmde.org.cn。
4、广东欧格斯科技有限公司对制氧机主动召回
东欧格斯科技有限公司生产的制氧机,生产批号OZ-5-01TW0,通过抽检发现,该批次个别产品由于员工在插制氧机磁阀端口线不牢靠,在运输途中可能导致端口线松脱,导致氧浓度低,广东欧格斯科技有限公司决定发起主动召回。
广东欧格斯科技有限公司对其生产的制氧机(注册证号:粤械注准20172080401 生产批号:OZ-5-01TW0)主动召回。召回级别为三级。涉及产品的型号、规格及批次等详细信息见《医疗器械召回事件报告表》。
5、国家药监局关于发布国家医疗器械监督抽检结果的通告(2024年第29号)
为加强医疗器械监督管理,保障医疗器械产品质量安全有效,国家药品监督管理局组织对心电图机、软性接触镜、幽门螺杆菌抗体检测试剂等10个品种进行了产品质量监督抽检,有13批(台)产品不符合标准规定。具体情况通告如下:
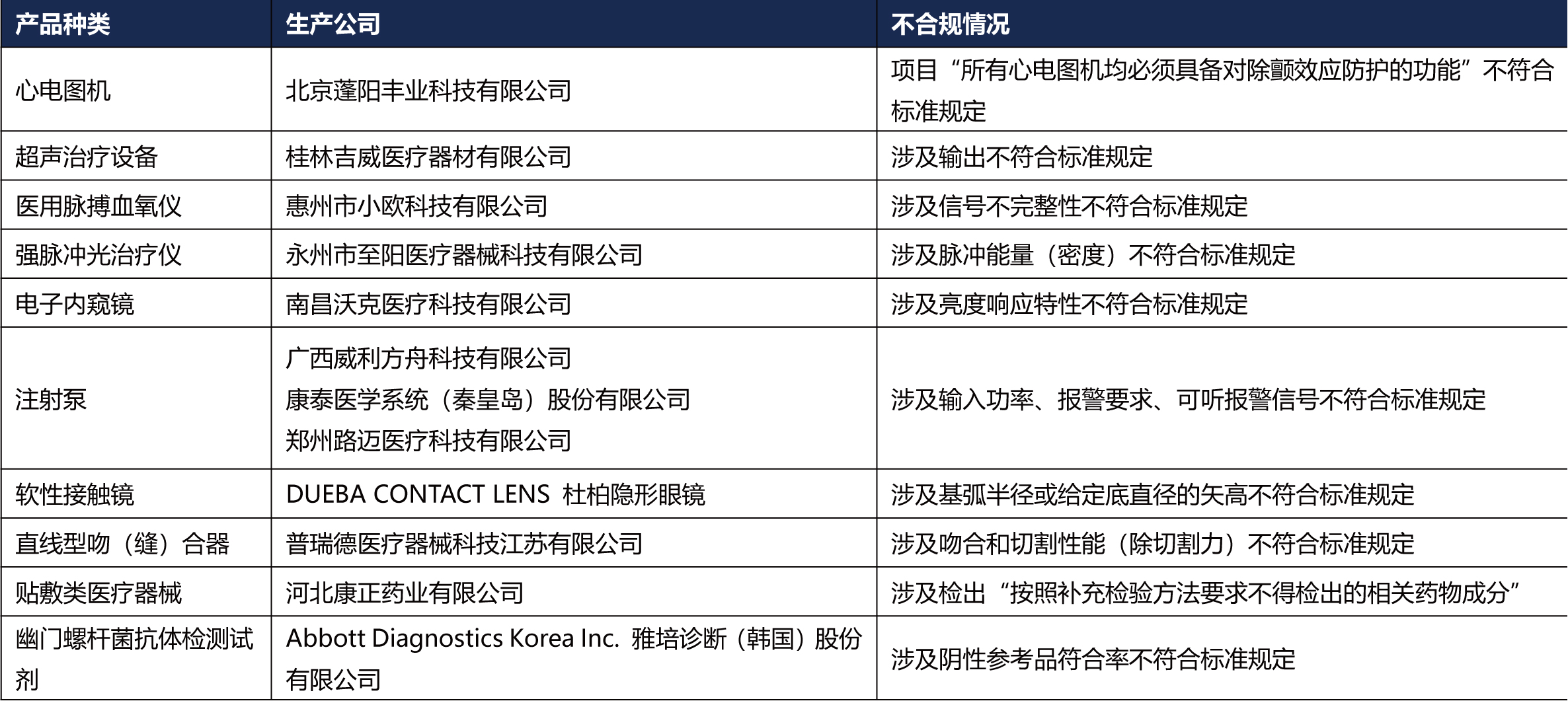
对这些产品,国家药品监督管理局已要求相关企业所在地省级药品监督管理部门按照《医疗器械监督管理条例》《医疗器械生产监督管理办法》《医疗器械召回管理办法》等要求,及时作出行政处理决定并向社会公布。省级药品监督管理部门要督促企业对抽检不符合标准规定产品进行风险评估,根据医疗器械缺陷的严重程度确定召回级别,主动召回产品并公开召回信息;督促企业尽快查明产品不合格原因,制定整改措施并按期整改到位。
6、生物可吸收雷帕霉素洗脱冠脉支架系统获批上市
近日,国家药品监督管理局批准了上海微创医疗器械(集团)有限公司“生物可吸收雷帕霉素洗脱冠脉支架系统”创新产品注册申请。
该产品由药物支架和输送系统组成。支架由支架基体、显影标记、药物涂层三部分构成,支架基体材料为左旋聚乳酸(PLLA),在支架近远端各有一个显影标记物,雷帕霉素药物涂层喷涂于支架外表面单面。输送系统为快速交换式球囊扩张导管。该产品经辐照灭菌,一次性使用,改善冠状动脉腔内直径,适用于冠脉原发病变导致的缺血性心脏病患者。
该产品采用了单面药物涂层技术,通过点涂工艺仅在支架外表面涂敷药物,支架杆侧面及内表面无药物涂层,提高了药物利用率,有利于血管内皮化进程。
药品监督管理部门将加强该产品上市后监管,保护患者用械安全。
7、广东省药品监督管理局关于注销《医疗器械生产许可证》的通告(2024年第6期)
按照《医疗器械监督管理条例》《医疗器械生产监督管理办法》有关规定,根据企业申请,广东省药品监督管理局依法注销广州市兴世电子有限公司等14家企业的《医疗器械生产许可证》。详细内容见原文链接附件。
8、广东省药品监督管理局关于同意亚弘(东莞)电器有限公司等19家注册人主动注销《医疗器械注册证》的通告
亚弘(东莞)电器有限公司等19家注册人向广东省药品监督管理局主动提出注销产品注册证的申请。根据《医疗器械监督管理条例》的规定,现对该19家注册人持有的22张《医疗器械注册证》依法予以注销(注销清单见附件)。
9、单间室膝关节假体直接参照全膝关节假体磨损测试方法ISO 14243进行磨损试验是否可行
目前我国尚无专门针对单间室膝关节假体的磨损试验方法的国家标准或行业标准,申请人可参考全膝关节假体磨损试验方法的国际标准,如ISO 14243-1或ISO 14243-3、ISO 14243-2等标准。建议结合单间室膝关节假体与全膝关节假体在临床使用情况上的差异进行合理调整和优化,对单间室膝关节假体磨损性能的可接受性进行合理性论述。假体磨损测试方法ISO 14243进行磨损试验是否可行
10、血液透析器产品的生物相容性评价项目至少包括哪些
建议参考GB/T 16886.1标准要求,至少包括细胞毒性、致敏反应、刺激或皮内反应、材料介导的致热性、急性全身毒性、亚急性毒性、亚慢性毒性、慢性毒性、植入反应、血液相容性、遗传毒性、致癌性(如适用)。
11、FDA发布《无菌类器械 (510(k))申报资料中无菌证明资料递交及审查》指南——器审中心官方解读
生物医用材料是诊断、治疗、修复或替换人体组织或器官,或增进其功能的一类高技术新材料。3D打印技术主要是以数字模型文件为基础,根据零件或物体的三维模型数据,通过成型设备以材料累加的方式制成实物模型的技术,在高精度、个性化制造及复杂形状构建等方面具有独特优势。但是3D打印医疗器械由于其外形设计的不确定性,针对每个设计进行完整的台架试验是不现实的,也缺乏合理性,因而需要建立一种替代验证方法。器审中心于2020年发布的《定制式个性化骨植入物等效性模型注册技术审查指导原则》给出了等效性模型的定义,并对等效模型建立的要求和方法进行了深入阐述。器审中心结合等效性模型在生物医用3D打印骨科植入物中的应用来进一步说明等效模型的构建要求及方法,以供参考,详细内容请见原文。
 上一篇
上一篇
服务赋能 协同创新 | 医疗器械产业质量提升交流会圆满举行
由华测纽唯发起的【广州开发区广州市黄埔区医疗器械行业质量基础设施服务工作站】于2024年5月10日15:00召开了黄埔区医疗器械行业质量提升交流会。
2024-05-20 00:51:42
医疗器械领域再获新突破!| 华测检测CMA扩项获证成功!
2024年2月27日,CTI华测检测有源医疗器械实验室(以下简称“华测医疗器械实验室”)顺利通过国家级CMA资质扩项评审,并已获得资质认定证书及扩项附表。
2024-03-04 00:43:27
CTI华测检测成功入围《深圳市医疗器械优质供应商推荐目录》
深圳市市场监督管理局为贯彻落实深圳市市委、市政府关于产业质量提升的工作部署,着力提升深圳市高端医疗器械产业质量竞争力,推动产业高质量发展,2023年4月11日,深圳市启动了医疗器械产业质量提升“灵渠”计划。
2024-01-22 06:37:44



 一键下单 流程透明
一键下单 流程透明 专业服务 权威公正
专业服务 权威公正 传递信任 彰显品质
传递信任 彰显品质 根植中国 服务世界
根植中国 服务世界
- 热线电话
- 业务咨询
- 快速询价
- 在线客服
- 报告验证